Gas chromatography (GC)
Determination of Benzene, Toluene and m-, p-, o-Xylene in water
Gas chromatography is one of the most popular chromatographic methods. It is commonly used for the separation of non-polar compounds with high vapor pressure and a vaporization without decomposition.
In this part of the practice course, you will learn fundamental chromatographic terms which will be experimentally determined by GC. We will analyze the amount of BTX (Benzene, Toluene, m-, p-, o-Xylene) in an unknown water sample. To extract the analytes, we will use liquid/liquid-extraction.
Methods
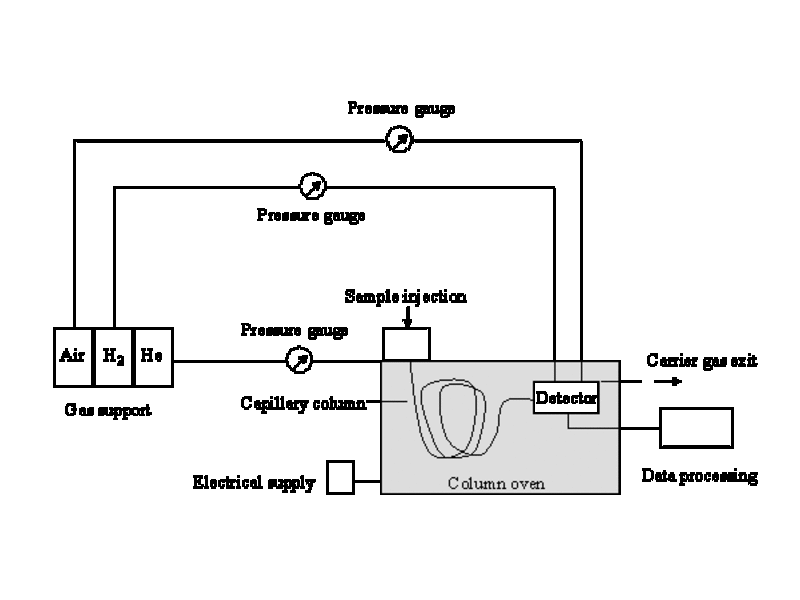
The sample is injected into the hot injector where it is vaporized. The carrier gas (in most cases: He) transports the vaporized sample through the thermostated capillary column into the detector, where the separated analytes are obtained as peaks in a chromatogram.
A gas chromatographic system consists of following parts: Injector – column – detector
Injector
The primary goal for any injection is to introduce the sample into the column. Liquid samples will be injected with small glass-syringes (volume 1–10µL for capillary columns and 50µL for packed columns). Gaseous samples can be injected with gas-tight glass-syringes with volumes between 1 and 10mL. A better way is injecting gaseous samples with a sample loop of a defined volume.
In all cases, the injection has to be reproducible without disturbation of the equilibrium state in the column, the sample has to be vaporized in a small zone, and must produce narrow peaks for every compound.
There are four types of injectors: split, splitless, megabore and "on-column". The first two types use a common injector called "split/splitless injector". Each type of injector is well suited for a particular type of sample.
A split/splitless injector consists of a glass tube ("liner") which creates an inert environment inside the injector and where the sample is vaporized. A syringe is used to pierce the septa and to introduce the sample into the injector. The injector temperature should be high enough to ensure instant vaporization without degradation of the sample. For most samples, 200-250°C as injector temperature is high enough. The sample vapor is mixed with the carrier gas and is transported into the column. Components of the sample that are not vaporized remain in the injector. The septum purge is a low flow which minimizes the amount of septum bleed materials which could contaminate the GC system. Septum purge gas sweeps the bottom of the septum and the top of the liner (labeled "T" for top at the GC) out through the purge vent. A typical septum purge flow is between 0.5 and 5 mL/min.
Split injection is a most common technique which is used for highly concentrated samples (concentrations between 0.1 and 10 µg/µL per single component). In this technique the sample is vaporized immediately, mixed with carrier gas and a small amount enters the column. The remaining sample leaves the injector via the split vent at the bottom of the liner (labeled "B" for bottom at the GC). Split ratios (resp.: "split") between 1:10 and 1:100 are typical.
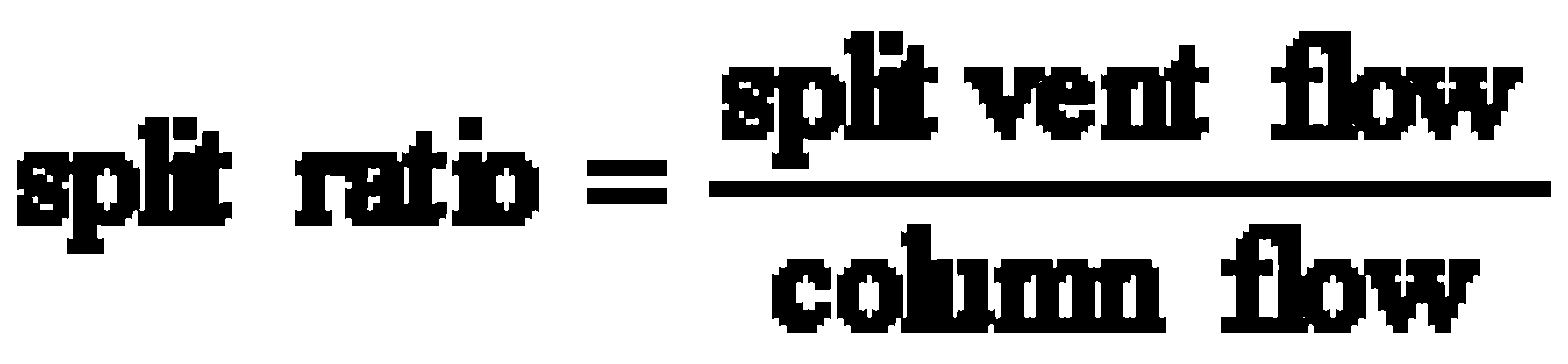
E.g.: Column flow = 2 mL/min
Split vent flow = 100 mL/min
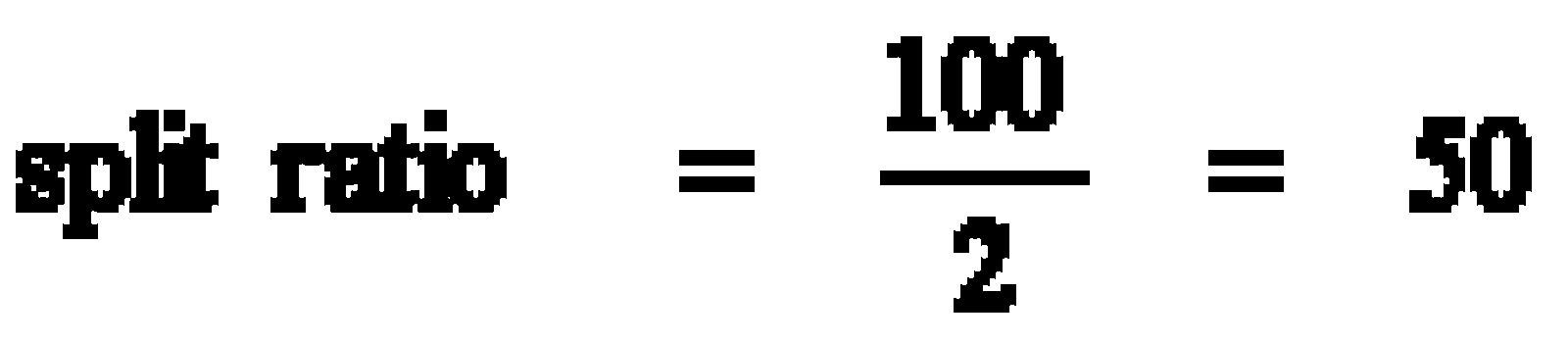
split = 1:50
Splitless injection is used with a closed split flow for lower concentrated samples.
Megabore direct injection is used with packed columns (0.45 and 0.53 mm I.D.).
The on-column injection is not a vaporization technique. The sample is deposited directly into the column without vaporization. It is used for high boiling compounds like petroleum waxes or thermally labile compounds.
Column
The separation takes place in the column by physico-chemical interaction of the sample compounds with the stationary and the mobile phases. The separation depends on stationary phase type, stationary film thickness and column inner diameter (factors which are interrelated), and column length. So the capillary column must be selected deliberately. The selectivity is influenced by polarizability, solubility, magnitude of dipoles and hydrogen bonding behavior of the stationary phase with the different compounds.
The stationary phase determines the ability of the column to separate sample components. If the stationary phase retains one compound to a greater extent than another, the compounds can be separated. The selection of the stationary phase is based on the following chemical principle: a non polar column is better for separation of non polar analytes, and polar columns effectively separate polar analytes. The polarity of the stationary phase is determined by the structure of the polymer which constitutes the stationary phase. A non polar column (stationary phase: e.g. polydimethylene-siloxane) separates the analytes regarding to their boiling points and their different vapor pressure. With particulate substitution of methylene groups in the stationary phase by phenyl- or cyanopropyl groups, the polarity of the stationary phase increases. High polar stationary phases are based on polyethylene glycols. In this case, the separation takes place due to the polarity and the boiling points of the compounds.
Two kinds of columns are available: capillary columns and packed columns. Both have an adsorbens, and their surfaces are covered with a liquid film as stationary phase. Packed columns are completely filled with the adsorbens whereas capillary columns – which are mostly used for GC analysis - are open-tubular columns. They are composed of three parts: a glass or fused silica tube (1.) covered with the stationary phase (2.), and the outer surface consisting of a polyimide coating (3.), which protects the column from breaking.
The film thickness of the stationary phase directly influences the retentive character and capacity of a column. Increasing film thickness leads to an increase in solute retention. The capacity of a column is defined as the maximum amount of a compound, which can be injected into a column before peak distortion occurs. Capacity is directly related to film thickness, diameter and stationary phase polarity. Increased capacity is a result of increasing diameter and film thickness. The more soluble a solute in the stationary phase the greater the capacity for the solute.
There are two options of stationary phase film thickness. Thick film columns (film thickness: 1–5µm) are especially suited for analytes with low boiling points (e.g., volatile organics and gases). Thin film columns (film thickness: 0,10–0,25µm) are better for analytes with high boiling points, low volatility or simple samples. The analytes elute at lower temperature and in better time.
The column diameter has a direct impact on the efficiency, retention times and sample's capacity of the column. Smaller diameter columns have a greater efficiency than larger diameter columns, which have instead a greater sample capacity. The inner diameter of capillary columns ranges between 0,15mm and 0,53mm.
The retention time (tR) is directly influenced by the column length. Longer columns cause higher retention times. Capillary column length ranges between 10m up to 100m, but commonly 25m or 60m columns are used.
The column temperature influences the separation, too. An increased temperature induces a decrease of the distribution coefficient and the retention time. Simultaneously the viscosity of the carrier gas increases, which leads to an increasing dead time and a decreasing gas flow.
Detector
The detector converts analytes into an electrical signal which should be linear depending on the amount or concentration of the analytes. All measurements must be done in the linear range of the detector. The quantification of the compounds could be done by a multi-point calibration. For all detectors, one or more gases are necessary like combustion, reagent, auxiliary and make-up gases.
The Flame ionization detector (FID) indicates compounds with C-H-bondings. The sample is burned in an H2/air-flame and the produced ions change the electrical conductivity. The electrical conductivity of the H2/air-flame is low due to low ionization. Organic molecules usually burn with formation of ions and liberation of electrons, thus increasing the electrical conductivity of the flame.
The Electron capture detector (ECD) indicates electronegative compounds which catch electrons (emitted from 63Ni) and thereby reduce the current. The ECD is more sensitive than the FID, so it is used for samples with low analyte concentrations.
| Detector | Selectivity | Sensitivity | Linear range | Gases | Temperature |
|---|---|---|---|---|---|
| FID | C-H | 0.1-10ng | 107 | combustion: H2 and air make-up: He or N2 | 250-300°C |
| ECD | Halogens, nitrates and conjugated carbonyls | 0.1-10pg (halogens); 1-100pg nitrates; 0.1-1ng (carbonyls) | 104 | N2 or Ar/CH4 | 300-400°C |
| NPD | N and P | 1-10pg | 106 | combustion: H2 and air make-up: He | 250-300°C |
| FPD | S and P | 10-100pg (S); 1-10pg (P) | non-linear (S); 104 (P) | combustion: H2 and air make-up: N2 | 250-300°C |
| TCD | all compounds except the carrier gas | 5-20ng | 105 | make-up: like carrier gas | 150-250°C |
| PID | Usually aromatics and olefins | 25-50pg (aromatics); 50-200pg (olefins | 107 | make-up: like carrier gas | 200°C |
| MSD | any compound which fragments within the select mass range | 1-10ng (full scan); 1-10pg (SIM) | 105 | none (vacuum) | 250-300°C (transfer line); 150-250°C (source) |
NPD: nitrogen-phosphorous detector, FPD: Flame photometric detector, TCD: thermal conductivity detector, PID: Photoionization detector, MS: mass selective detector
Detection of Benzene, Toluene and m-, p-, o-Xylene (BTEX) in unknown water
Material
- Gas chromatograph with flame ionization detector (FID)
- Graduated pipette, volumetric pipette (Eppendorf) of different volumes
- Centrifuge tubes
- Sample vials, septa, and tong
- n-Hexane (flammable, irritant; R: 11-48/20) for trace analysis
- 10 mL volumetric flasks
- Sodiumsulfate
- Ethylbenzene-single standard (0.8 mg/mL)
- Single standards (without exact concentration specification) of Benzene, Toluene, m-Xylene, p-Xylene, o-Xylene (about 10 drops in 800 µL n-hexane)
- BTX-mixture in n-hexane:
- Benzene (0.8 mg/mL)
- Toluene (0.8 mg/mL)
- meta-Xylene (0.8 mg/mL)
- para-Xylene (0.8 mg/mL)
- ortho-Xylene (0.8 mg/mL)
Purpose

Analyze the amount of BTX in an unknown water sample. Identification of the analytes, followed by a quantification with internal and external standard using a 4-point-calibration in the range of 0 to 40µg/mL (40 ppm) has to be done.
Methods
To perform organic trace analysis, it is necessary to clean all used flasks, bottles and all other equipment with acetone and/or n-hexane to remove any contamination. Usually glass vessels are used.
Execution
1. Determine the dead time of the system by injection of methane (unretained compound). Calculate the linear velocity (mL/min) of the carrier gas (He) and the average carrier gas flow (cm/s) from the measured retention time (tM) for methane using the equations below.
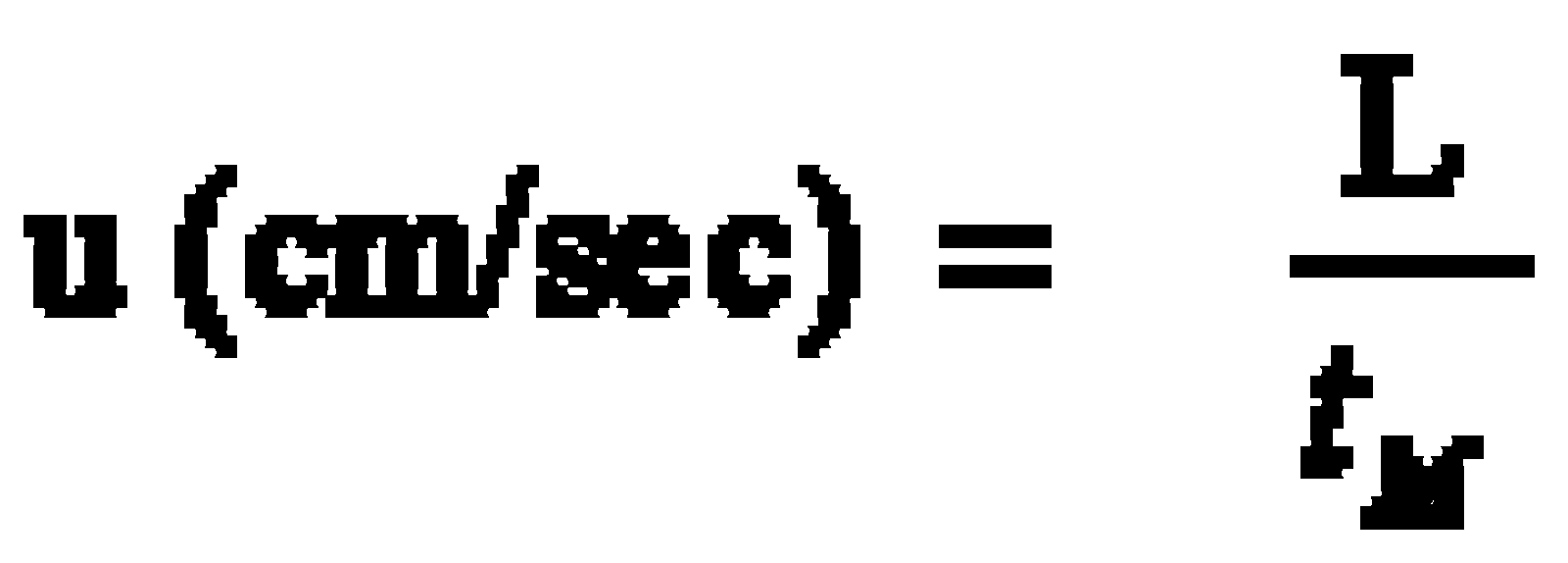
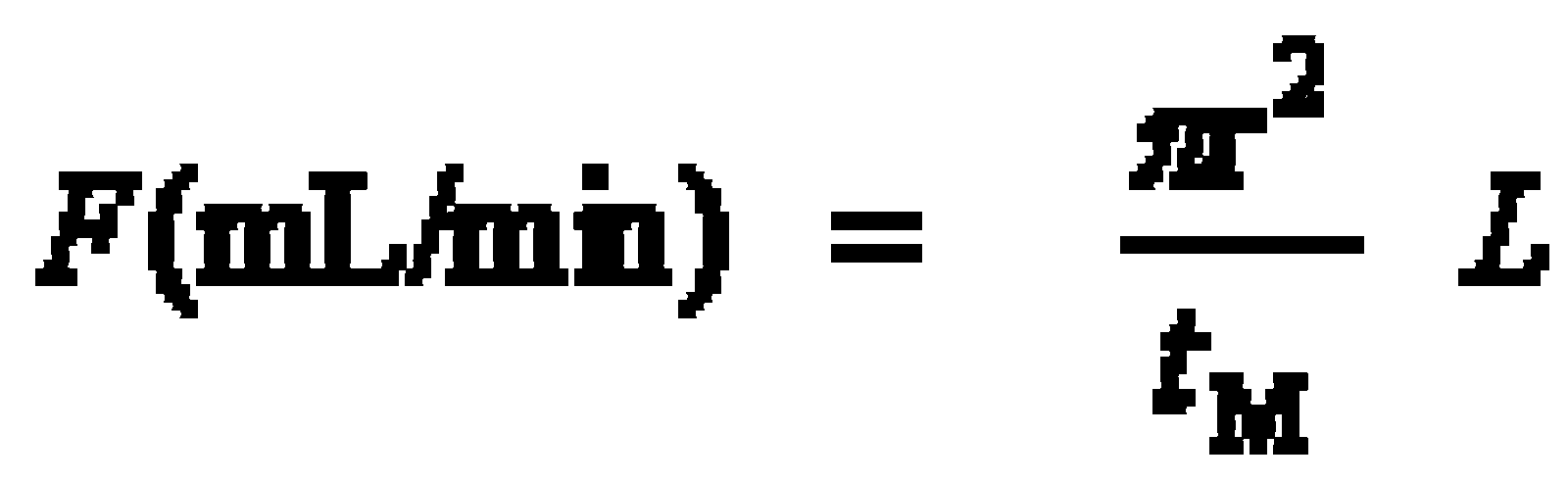
u Average linear velocity (cm/sec)
L Column length (cm)
tM Retention time of a unretained compound (Eq. 1:sec; Eq. 2: min)
F Average volumetric flow rate (mL/min)
r Column radius (cm)
2. Subsequently, set the head pressure (He), the split (B, bottom) and the purge (T, top) to get the following GC-conditions:
| column | OPTIMA 5 DF, 60m ´ 0.25 mm, 0.25 µm film |
|---|---|
| temperature program | 40 °C (5), 2°C/min, 48°C, 15°C/min, 120°C |
| FID- temperature | 250 °C |
| injector temperature | 250 °C |
| carrier flow | 1 mL/min |
| head pressure | 175 kPa |
| purge | 1 mL/min |
| H2 pressure | 60 kPa |
| air pressure | 120 kPa |
| injection volume | 1 µL |
3. Determine the retention times of Benzene, Toluene, o-, m-, p-Xylene and Ethylbenzene by single injection.
4. Analyze a blank sample (200 µL Ethylbenzene-single standard in 10 mL n-Hexane).
5. Calculate the retention time of Ethylbenzene, the capacity factor, the number and the height of the theoretical plates for the actual carrier gas velocity.
6. Calibration
7. Prepare 4 different dilutions of the BTX-mixture (BTX: 0.8mg/mL): Add 50 µL, 100 µL, 250 µL and 500µL of the standard mixture in 10mL n-hexane and add 200 µL of the Ethylbenzene-single standard (0.8 mg/mL) as internal standard to the four solutions.
Which BTX-concentrations have the obtained solutions ?
Fill approx. 2 mL of every solution in sample vial and close with a cup and analyse with the GC (calibration).


8. Determine the amount of BTX in an unknown water sample
9. Sample extraction (Liquid-liquid-Extraction):
Pour exactly 10mL of the sample (unknown water) in a centrifuge tube, add 40µL Ethylbenzene (single standard: 0.8 mg/mL) and 1mL n-Hexane. Close the tube with a buckler and shake it intensively for 2 minutes.
Wait until two phases separate (if necessary centrifuge) and carefully pipette the organic phase in a centrifuge tube with five or more portions (spoonful) of sodiumsulfate to dry the solution.
Add once again 1 mL n-hexane to the water sample, shake, pipette the organic phase in the same centrifuge tube, shake it and centrifuge. Then take the organic phase in a GC-vial and close it with the tong. Analyse the sample with the GC.
10. Calculate the recovery of the extraction method.
11. To determinate the recovery add 40 µL ethylene benzene (0.8 mg/mL) and 40µL of the BTX-mixture (0.8 mg/mL) to 10mL of the water sample (spiked water sample) and extract the water like the description above (see 7.).
Extraction scheme:
| sample no. | name | water sample | Ethylbenzene single standart | BTX mixture | n-Hexane |
|---|---|---|---|---|---|
| 1 | A1 | 10 mL | 40 µL | - | 2 x 1 mL |
| 2 | A2 | 10 mL | 40 µL | - | 2 x 1 mL |
| 3 | B1 | 10 mL | 40 µL | - | 1 x 2 mL |
| 4 | B2 | 10 mL | 40 µL | - | 1 x 2 mL |
| 5 | recovery1 | 10 mL | 40 µL | 40 µL | 2 x 1 mL |
| 6 | recovery2 | 10 mL | 40 µL | 40 µL | 1 x 2 mL |
12. Evaluation:
Plot the calibration curve (area versus concentration) and calculate the concentration of the unknown water sample and the spiked water sample (recovery 1 and 2). Compare evaluations with internal and with external standard. Determine the recovery of the method.